
Medical devices play an essential role in healthcare by providing diagnostic, preventive, therapeutic, monitoring, or rehabilitative functions. In Indonesia, medical devices are regulated by the Ministry of Health (MoH), which ensures their safety, efficacy, and quality. The regulatory framework categorizes medical devices into different classes based on risk, which dictates the regulatory process, registration requirements, and oversight. This article provides an in-depth look at these classifications, the criteria for each, and the associated registration process in Indonesia.
Classification of Medical Devices in Indonesia
Medical device classification in Indonesia is crucial in defining the regulatory framework for various devices based on their associated risks. The system aligns with international guidelines, including the Global Harmonization Task Force (GHTF) and the ASEAN Medical Devices Directive (AMDD), to ensure global harmonization while accommodating local needs. This classification into Class I, II, and III is not just a categorization but a foundational aspect influencing the entire regulatory process, including the complexity of registration, the scrutiny applied, and associated costs. Each classification dictates specific regulatory requirements, ensuring that devices are appropriately evaluated for safety and efficacy based on their risk profile.
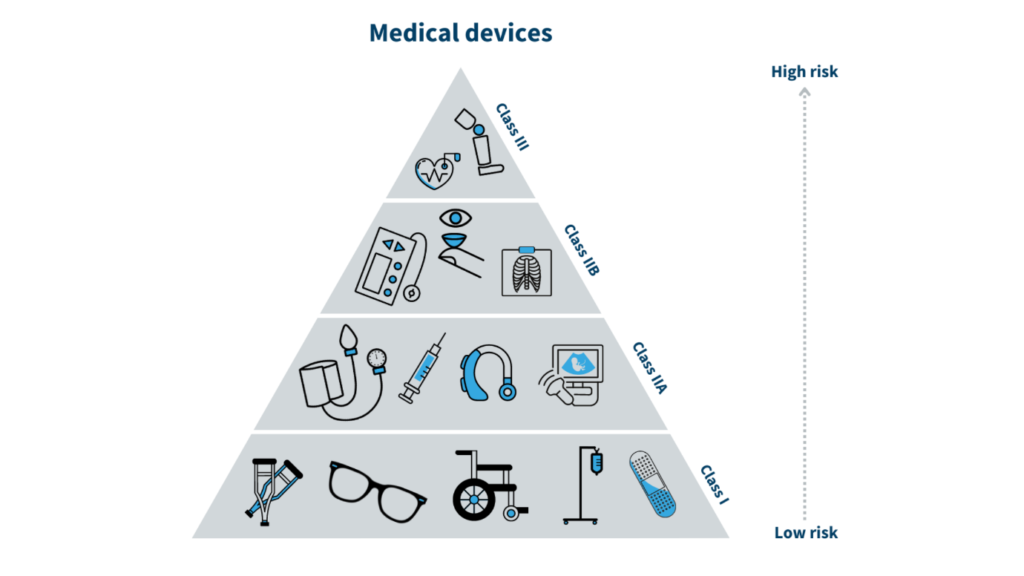
Class I (Low-Risk Devices)
Class I devices are the least complex among the three classifications, indicating a minimal risk to the user. These devices do not generally require sterile conditions or special monitoring to be safe and effective. Common examples of Class I devices include simple bandages, non-measuring stethoscopes, crutches, and surgical gloves. The criteria for this classification focus on the low-risk profile of these devices. They are non-invasive, meaning they do not enter the body or penetrate the skin and do not require direct measurements for proper functioning. The regulatory requirements for Class I devices are relatively straightforward. The registration process is streamlined, typically taking about 15 days, and involves minimal documentation, which includes device labeling, manufacturer information, and a basic risk analysis. The associated fees for registering Class I devices are also lower, amounting to approximately IDR 1,500,000 (US$115).
Class II (Moderate-Risk Devices)
Moving to Class II, the devices are considered to pose a higher risk compared to Class I, necessitating more stringent regulatory oversight. These devices include infusion pumps, ECG machines, X-ray machines, and anesthesia machines. The criteria for Class II devices reflect their moderate health risk, as they are invasive or may come into direct contact with users, necessitating careful monitoring and sometimes sterilization. The regulatory requirements for Class II devices include a more comprehensive registration process, typically taking around 30 days. This process involves a detailed assessment that includes clinical evaluation reports, risk management documents, and a Good Manufacturing Practice (GMP) certificate. The associated fees are higher, around IDR 3,000,000 (US$230), reflecting these devices’ increased scrutiny and regulatory demands.
Class III (High-Risk Devices)
Class III represents the highest risk category, reserved for devices that could cause serious injury or death if they malfunction. These include implanted devices such as pacemakers, defibrillators, and prosthetics. The criteria for Class III devices focus on their high-risk nature, as they are typically implantable or provide critical measurements necessary for therapeutic decisions. The regulatory requirements for these devices are the most rigorous, with the registration process taking up to 45 days. This category demands comprehensive documentation, including clinical trials, biocompatibility studies, and a clinical evaluation report. The registration fees for Class III devices are also the highest, around IDR 5,000,000 (US$340), reflecting the critical importance of these devices to patient safety. The stringent requirements ensure that only devices with proven safety and efficacy are approved for use in Indonesia.
The Registration Process for Medical Devices in Indonesia
The registration process for medical devices in Indonesia involves a multi-step procedure designed to ensure these devices’ safety, efficacy, and quality before they can be legally marketed. The process varies according to the device’s classification (Class I, II, or III), and each has its own requirements. Understanding these steps is crucial for manufacturers looking to enter the Indonesian market, as they dictate the documentation needed and the timeline and costs associated with each device category.
Pre-market Registration
The process begins with submitting an application form to the Ministry of Health (MoH) through a licensed local importer or distributor. This form must include comprehensive information about the device, such as its intended use, design specifications, and clinical evaluations. The applicant must provide an executive summary outlining the device’s purpose and benefits and detailed labeling information, including instructions for use, contraindications, warnings, and precautions. Additionally, the submission requires manufacturer details, including ISO 13485 certification, a standard for quality management in medical devices, demonstrating compliance with international best practices in manufacturing. A risk analysis is crucial in evaluating potential risks associated with the device’s use. A clinical evaluation report based on clinical trials and studies showing safety and efficacy is also necessary. Moreover, a Free Sales Certificate (FSC) from the manufacturer’s country, certifying that the device is freely sold there, is required to demonstrate the device’s acceptance in international markets. Biocompatibility testing documentation must be provided, showing that the device’s materials are safe for human use. The registration fees vary by device classification; Class I devices require a lower fee of around IDR 1,500,000 (US$115). Class II and III devices involve higher costs due to the increased complexity and documentation requirements.
Post-market Registration
After the MoH evaluates all submitted documents and conducts its risk assessment, a product license, and a registration number are issued. The license holder must be a local entity, either an importer or distributor, who must maintain the registration. The validity of the product license typically ranges from 2 to 5 years, depending on the type of device and the Letter of Authorization (LOA). Renewal of the license is necessary before its expiration to continue marketing the device legally in Indonesia. This post-market phase is crucial for maintaining compliance with regulatory standards, especially if there are changes in the device’s packaging, labeling, or technical specifications. An amendment application can be submitted for minor changes, whereas major modifications might necessitate a new registration. It is the responsibility of the local entity to ensure that all updates and amendments are promptly communicated to the MoH, maintaining the device’s compliance throughout its lifecycle in the market. This regulatory oversight post-registration is designed to ensure continuous safety and efficacy, allowing the MoH to monitor adverse events and make necessary updates to the product specifications as needed.
E-Catalogue Registration in Indonesia
The Indonesian government’s introduction of the e-Catalogue system marks a pivotal shift in how medical devices are registered and procured. This online platform allows registered medical devices to be listed and accessible to healthcare providers and government purchasing entities, thereby streamlining the procurement process. Including a device in the e-Catalogue involves a thorough evaluation to ensure it meets specific standards and regulatory requirements set by the Ministry of Health (MoH). After obtaining a product license, the device can be uploaded to the e-Catalogue through a licensed local importer. This process requires submitting necessary documentation, such as executive summaries, detailed labeling information, risk analyses, and clinical evaluation reports. To ensure safety and efficacy, the device must comply with local quality management standards, including ISO 13485 certification.

The benefits of listing a device in the e-Catalogue are manifold. It reduces administrative burdens for manufacturers and healthcare providers by consolidating procurement processes into a single digital platform. This speeds up the purchasing process and lowers procurement costs through increased supplier competition. Moreover, the e-Catalogue system enhances transparency, allowing for easier comparison of devices based on their technical specifications, pricing, and safety records. For manufacturers, being listed in the e-Catalogue can significantly increase market access and visibility, especially for foreign entities. It provides a centralized location where government institutions can quickly identify and procure necessary devices, thus improving overall healthcare delivery in Indonesia.
Challenges and Considerations for Medical Device Registration in Indonesia
Despite its advantages, the e-Catalogue system also introduces challenges that manufacturers must navigate. One of the primary hurdles is the requirement for foreign manufacturers to work through a licensed local importer to submit their application for registration. This intermediary role can add additional costs and complexities to the process. The involvement of a local representative ensures compliance with local regulations. It facilitates communication with the MoH but can also lead to delays if the importer’s understanding of regulatory requirements is lacking or insufficient coordination between the parties involved. Communication with the MoH is another critical challenge. The regulatory process may involve several interactions where additional information or corrections are requested. This can be a bottleneck if the documentation submitted is incomplete or there are ambiguities in the provided data. The iterative nature of the communication process could delay the registration timeline, impacting the availability of the device in the market. Furthermore, the regulatory framework in Indonesia is dynamic, with continuous updates, such as the introduction of Halal certification requirements for medical devices. Manufacturers must remain vigilant and informed about any changes to the regulatory landscape to ensure their products remain compliant. Keeping abreast of these updates requires active engagement with the MoH and proactive management of the regulatory portfolio, which can be resource-intensive, particularly for foreign companies unfamiliar with the local regulatory environment.
Conclusion
In conclusion, the classification of medical devices in Indonesia follows a structured framework aligned with global standards, specifically the Global Harmonization Task Force (GHTF) guidelines, and harmonized with the ASEAN Medical Devices Directive (AMDD). Class I, II, and III classification determines the level of risk the device poses to patients and users, influencing the registration process, regulatory scrutiny, and associated costs. Each class has distinct criteria and regulatory requirements, from minimal documentation for Class I devices to comprehensive evaluations for Class III devices, including clinical trials and biocompatibility testing. The introduction of the e-Catalogue further streamlines procurement, enhancing transparency and efficiency. Navigating these classifications and registration processes requires a thorough understanding of both the regulatory landscape and the specific demands of the e-Catalogue system, which is crucial for manufacturers aiming to succeed in the Indonesian market.

