
医療機器は、診断、予防、治療、モニタリング、リハビリテーションなどの機能を提供することで、医療において重要な役割を果たしています。インドネシアでは、医療機器は保健省(MoH)によって規制されており、その安全性、有効性、品質が保証されています。この規制枠組みでは、医療機器をリスクに基づいて様々なクラスに分類し、規制プロセス、登録要件、監督を規定しています。この記事では、これらの分類、それぞれの基準、そしてインドネシアにおける関連する登録プロセスについて詳しく説明します。
インドネシアにおける医療機器の分類
医療機器 インドネシアにおける医療機器のクラス分けは、様々な医療機器の関連リスクに基づく規制枠組みを定義する上で極めて重要です。このシステムは、GHTF(Global Harmonization Task Force)やAMDD(ASEAN Medical Devices Directive)などの国際ガイドラインに準拠しており、地域のニーズに対応しながら国際的な調和を確保しています。クラスI、II、IIIへの分類は単なる分類ではなく、登録の複雑さ、適用される審査、関連コストなど、規制プロセス全体に影響を及ぼす基本的な要素です。各分類には具体的な規制要件が規定されており、医療機器のリスクプロファイルに基づいて安全性と有効性が適切に評価されることが保証されています。
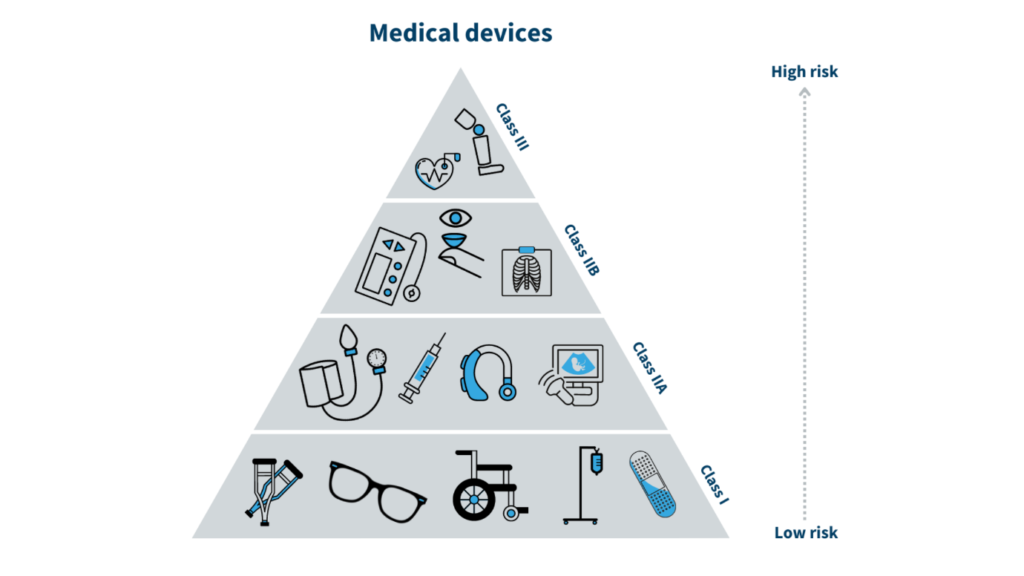
クラスI(低リスクデバイス)
クラス I 機器は 3 つの分類の中で最も単純で、ユーザーに対するリスクが最小限であることを示します。これらの機器は一般に、安全かつ有効であるために滅菌状態や特別なモニタリングを必要としません。クラス I 機器の一般的な例としては、単純な包帯、非測定聴診器、松葉杖、手術用手袋などがあります。この分類の基準は、これらの機器の低リスク プロファイルに重点を置いています。これらは非侵襲性であり、体内に入ったり皮膚を貫通したりすることはなく、適切に機能するために直接測定する必要はありません。クラス I 機器の規制要件は比較的簡単です。登録プロセスは合理化されており、通常約 15 日かかり、機器のラベル、製造元情報、基本的なリスク分析などの最小限の文書化が必要です。クラス I 機器の登録に関連する料金も低く、約 IDR 1,500,000 (US$115) です。
クラスII(中等度リスク機器)
クラスIIに移行すると、デバイスはクラスIと比較してより高いリスクをもたらすとみなされ、より厳格な規制監督が必要になります。これらのデバイスには、輸液ポンプ、心電図装置、X線装置、麻酔器が含まれます。クラスIIデバイスの基準は、侵襲性があるか、ユーザーと直接接触する可能性があるため、注意深い監視と場合によっては滅菌を必要とする、中程度の健康リスクを反映しています。クラスIIデバイスの規制要件には、通常約30日かかる、より包括的な登録プロセスが含まれます。このプロセスには、臨床評価レポート、リスク管理文書、適正製造基準(GMP)証明書を含む詳細な評価が含まれます。関連料金は、これらのデバイスに対する監視と規制要件の強化を反映して、約3,000,000 IDR(US$230)と高額です。
クラスIII(高リスク機器)
クラスIIIは最もリスクの高いカテゴリーで、故障した場合に重傷や死亡につながる可能性のある機器に使用されます。ペースメーカー、除細動器、義肢などの埋め込み型機器が含まれます。クラスIII機器の基準は、通常埋め込み型であるか、治療上の決定に必要な重要な測定値を提供するため、その高リスク性に重点を置いています。これらの機器に対する規制要件は最も厳格で、登録プロセスには最大45日かかります。このカテゴリでは、臨床試験、生体適合性試験、臨床評価レポートなどの包括的な文書化が求められます。クラスIII機器の登録料も最も高く、約5,000,000ルピア(US$340)と、患者の安全にとってこれらの機器が極めて重要であることを反映しています。厳格な要件により、安全性と有効性が実証された機器のみがインドネシアでの使用が承認されます。
インドネシアにおける医療機器の登録手続き
インドネシアにおける医療機器の登録プロセスは、医療機器が合法的に販売される前に、安全性、有効性、品質を確保するための複数のステップから構成されています。プロセスは機器の分類(クラスI、II、III)によって異なり、それぞれに独自の要件があります。これらのステップを理解することは、インドネシア市場への参入を目指すメーカーにとって非常に重要です。なぜなら、これらのステップによって、必要な書類、各機器カテゴリーに関連するスケジュール、そしてコストが決まるからです。
市場前登録
このプロセスは、認可を受けた現地輸入業者または販売業者を通じて保健省(MoH)に申請書を提出することから始まります。この申請書には、機器の用途、設計仕様、臨床評価など、機器に関する包括的な情報が記載されている必要があります。申請者は、機器の目的と利点を概説した概要と、使用方法、禁忌、警告、注意事項を含む詳細なラベル情報を提出する必要があります。さらに、申請には、医療機器の品質管理規格であるISO 13485認証など、製造業者に関する詳細情報も必要であり、製造における国際的なベストプラクティスへの準拠を実証する必要があります。機器の使用に伴う潜在的なリスクを評価するには、リスク分析が不可欠です。安全性と有効性を示す臨床試験および研究に基づく臨床評価レポートも必要です。さらに、機器が国際市場で受け入れられていることを証明するために、製造業者の国が発行する自由販売証明書(FSC)が必要です。機器の材料が人体への使用に安全であることを示す生体適合性試験の文書も提出する必要があります。登録料は機器の分類によって異なります。クラスIの機器は、約1,500,000ルピア(US$115)と低料金です。クラスIIおよびIIIの機器は、複雑さと書類提出の要件が増すため、費用が高くなります。
市販後登録
保健省が提出されたすべての書類を審査し、リスク評価を実施した後、製品ライセンスと登録番号が発行されます。ライセンス保有者は、輸入業者または販売業者のいずれかの現地法人でなければならず、登録を維持する必要があります。製品ライセンスの有効期間は、機器の種類と認可状(LOA)に応じて、通常2年から5年です。インドネシアで機器を合法的に販売し続けるには、ライセンスの有効期限前に更新する必要があります。この市販後段階は、特に機器の包装、ラベル、または技術仕様に変更があった場合、規制基準への適合を維持するために非常に重要です。軽微な変更については修正申請を提出できますが、大幅な変更には新規登録が必要になる場合があります。現地法人は、すべての更新と修正を速やかに保健省に通知し、市場での機器のライフサイクル全体を通じて適合性を維持する責任を負います。この登録後の規制監督は、継続的な安全性と有効性を確保するために設計されており、保健省は有害事象を監視し、必要に応じて製品仕様を更新することができます。
インドネシアにおける電子カタログ登録
インドネシア政府による電子カタログシステムの導入は、 医療機器 登録・調達が可能な医療機器は、このオンラインプラットフォームを通じて登録・調達されます。登録済みの医療機器は、医療提供者や政府調達機関がアクセスできるようリスト化され、調達プロセスが効率化されます。機器を電子カタログに掲載するには、保健省(MoH)が定める特定の基準と規制要件を満たしていることを確認するための徹底的な評価が必要です。製品ライセンスを取得後、認可を受けた現地輸入業者を通じて機器を電子カタログにアップロードできます。このプロセスでは、概要、詳細なラベル情報、リスク分析、臨床評価報告書などの必要書類を提出する必要があります。安全性と有効性を確保するため、機器はISO 13485認証を含む現地の品質管理基準に準拠している必要があります。

電子カタログへの機器掲載のメリットは多岐にわたります。調達プロセスを単一のデジタルプラットフォームに統合することで、メーカーと医療提供者の事務負担が軽減されます。これにより、購買プロセスが迅速化され、サプライヤー間の競争が激化することで調達コストが削減されます。さらに、電子カタログシステムは透明性を高め、技術仕様、価格、安全性記録に基づいた機器の比較を容易にします。メーカーにとって、電子カタログへの掲載は、特に外国企業にとって市場へのアクセスと認知度を大幅に向上させます。政府機関が必要な機器を迅速に特定・調達するための一元的な拠点となり、インドネシアにおける医療サービス全体の改善につながります。
インドネシアにおける医療機器登録の課題と検討事項
電子カタログシステムには多くの利点がある一方で、メーカーが乗り越えなければならない課題も存在します。主なハードルの一つは、外国メーカーが登録申請を行う際に、認可を受けた現地輸入業者を経由する必要があることです。この仲介役は、プロセスに追加のコストと複雑さをもたらす可能性があります。現地代理人の関与により、現地規制への準拠が確保されます。保健省とのコミュニケーションは円滑になりますが、輸入業者の規制要件に対する理解が不足している場合や、関係者間の調整が不十分な場合は、遅延につながる可能性があります。保健省とのコミュニケーションは、もう一つの重要な課題です。規制プロセスでは、追加情報や修正の依頼など、複数回のやり取りが必要になる場合があります。提出された書類が不完全であったり、提供されたデータに曖昧さがある場合、これがボトルネックとなる可能性があります。コミュニケーションプロセスの反復的な性質により、登録スケジュールが遅延し、市場での機器の入手可能性に影響を与える可能性があります。さらに、インドネシアの規制枠組みは動的であり、医療機器に対するハラール認証要件の導入など、継続的に更新されています。製造業者は、製品のコンプライアンスを維持するために、規制状況のあらゆる変更について常に注意を払い、情報を入手する必要があります。これらの最新情報を把握するには、保健省との積極的な連携と規制ポートフォリオの積極的な管理が求められますが、特に現地の規制環境に不慣れな外資系企業にとっては、多くのリソースを費やすことになる可能性があります。
結論
結論として、 医療機器 インドネシアの医療機器登録制度は、世界基準、特にGHTF(医療機器規制国際調和タスクフォース)のガイドラインに準拠し、ASEAN医療機器指令(AMDD)と調和した構造化されたフレームワークに従っています。クラスI、II、IIIの分類は、機器が患者とユーザーにもたらすリスクのレベルを決定し、登録プロセス、規制調査、関連コストに影響を与えます。各クラスには、クラスI機器に対する最小限の文書化から、臨床試験や生体適合性試験を含むクラスIII機器の包括的な評価まで、独自の基準と規制要件があります。電子カタログの導入により、調達がさらに合理化され、透明性と効率性が向上します。これらの分類と登録プロセスを進めるには、規制環境と電子カタログシステムの特定の要求の両方を完全に理解する必要があり、これはインドネシア市場での成功を目指すメーカーにとって非常に重要です。


